The Build Structure tool builds and modifies atomic structures. The equivalent commands are build, torsion, bond, and angle. See also: Rotamers, Modeller Comparative, Model Loops, Add Hydrogens, Altloc Explorer, Renumber Residues, Change Chain IDs, rna, swapaa, swapna, tug, undo, selection context menus
Build Structure can be opened from the Structure Editing section of the Tools menu and manipulated like other panels (more...). Sections:
The Start Structure section of Build Structure adds new atoms or molecules independent of any pre-existing atoms. See also: Fetch by ID, open
Options to Add:
Clicking Apply adds the atom(s) with the specified Residue name, in either an existing molecule model or a new model with a specified name.
Clicking Apply brings up another dialog for specifying backbone φ (phi) and ψ (psi) angles and other parameters. One or more rows can be chosen with the mouse and Set to values either entered manually or supplied for various types of secondary structure:
| description | φ (°) | ψ (°) |
|---|---|---|
| α helix | -57 | -47 |
| antiparallel β strand | -139 | 135 |
| parallel β strand | -119 | 113 |
| 310 helix | -49 | -26 |
| π helix | -57 | -70 |
A block of rows can be chosen by dragging, or by clicking on the first (or last) line in the desired block and then Shift-clicking on its last (or first) line.
Sidechain conformations will be taken from the specified Rotamer library (see swapaa for literature citations):
The rotamer at each position will be chosen as described for the command swapaa with the criteria cp, meaning the rotamer with the fewest clashes, and if a tie, then the highest probability according to the rotamer library. The residues are added in N→C order, so only clashes with more N-terminal residues are evaluated. The peptide will be assigned the specified chain ID.
Clicking OK dismisses the dialog and creates the peptide. Hydrogen atoms are not included. Backbone bond lengths and angles are taken from the Amber ff99 parameters. Sidechain bond lengths and angles are taken from the Amber parameter files all*94.lib.
The Modify Structure section of Build Structure can change the element, valence (number of directly attached atoms), and/or geometry (spatial arrangement of attached atoms) of a single selected atom at a time, potentially changing its atom type assignment. Hydrogens are appended as needed to fill the valence. Building outward can be done by successive cycles of modifying a hydrogen attached to the previously modified atom.
Clicking Apply changes the selected atom as specified:
There is no checking as to whether the specified number of bonds and geometry are chemically reasonable. Hydrogens will be added to the atom to generate the indicated total number of bonds. They are added to form the idealized bond angles for the specified geometry, or if the atom already has two or more substituents, to maximally avoid those substituents. The default geometry is the same as for the atom's current type. Since the geometry around the atom may be changing, any pre-existing directly attached hydrogens are removed beforehand. No other atoms are removed automatically. If the atom is already bonded to one (and only one) other nonhydrogen atom, the bond will be adjusted to an approximate length depending on the elements involved. No other atoms are moved. Bond lengths for X-H (X = C/N/O/S) are taken from the Amber parm99 parameters.
In PDB format, an atom name can be up to four characters long and should be unique within a residue. An atom name normally starts with the element symbol.
In PDB format, residue names are normally three characters long and chain identifiers a single character.
Clicking the Delete button at the bottom of Modify Structure removes any selected atoms and bonds. See also command: delete, menu: Actions... Delete
The Adjust Bonds section of Build Structure allows adding and deleting covalent bonds. A bond can also be deleted using its selection context menu. See also: bond, combine, delete
The Adjust Torsions section of Build Structure
is a table of active (rotatable) torsions with entry fields and dials
for interactive adjustment. Clicking Activate adds the
selected bond to the list.
A torsion can also be activated using the
selection context menu of a bond.
An error message will appear if an attempt is made to activate a bond that is
terminal (without further atoms attached) or within a ring.
Active torsions are saved in
sessions.
See also:
Angles/Torsions,
angle,
torsion, the
bond rotation mouse mode
![]()
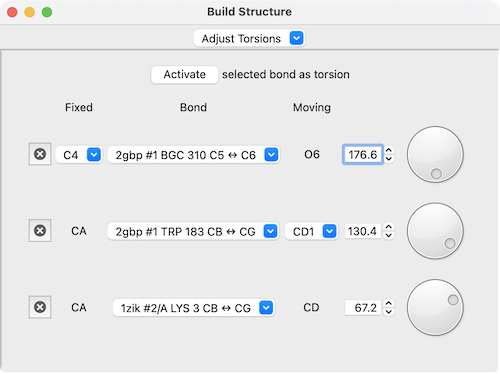 |
If the four atoms defining a torsion are called 1-2-3-4, atom 1 is Fixed and atom 4 is Moving, meaning that 4 and the side of the molecule bonded to 4 will rotate when the bond is rotated. The bond can be rotated by entering a new angle value or by manipulating the associated dial.
The Bond column contains a pulldown menu for each torsion labeled with the identifiers of atoms 2 ↔ 3 (those flanking the rotatable bond). This menu contains choices to:
When atom 2 is bonded to more than two atoms, there is more than one possible Fixed atom to define the measurement, and alternatives (if any) are available from a pulldown menu. Similarly, when atom 3 is bonded to more than two atoms, there is more than one possible Moving atom, and alternatives (if any) are available from a pulldown menu.
The torsion can be deactivated by clicking the “x” button to its left. To preserve the original position, make sure to reset the angle before deactivating it or swapping sides.
The Join Models section of Build Structure allows forming a covalent bond between two atomic models to combine them into one. The atoms of one model are repositioned and incorporated into the other model; the original model will no longer exist. Thus, it may be a good idea to save a session beforehand, since the join (along with other building actions) cannot be undone. The related command combine also combines atomic models, but without forming a bond or changing the relative positions of the atoms.
The atoms to be bonded must first be selected.
For Peptide Bond:
The selection must include exactly one peptide C-terminal carbon atom C –OR– N-terminal nitrogen atom N (not both) from one model, and from a second model, exactly one of whichever of those two atoms is not in the first. These C and N atoms each must be bonded to only one carbon (except N in proline or hydroxyproline can be bonded to two carbons); however, it may also be bonded to hydrogen and/or OXT, and if so, these atoms will be replaced as appropriate by the new peptide bond. The peptide bond will be added with the specified parameters:For Other Bond (the more general case):The values of ω (omega), φ (phi), and other peptide torsion angles are attributes of the amino acid residue containing the newly bonded N.
- C-N length (default 1.330 Å)
- Cα-C-N-Cα dihedral (ω angle) (default 180.0°)
- C-N-Cα-C dihedral (φ angle) (default –120.0°)
- Move atoms in [ selected N atom / selected C atom / smaller / larger ] model – which of the two models to reposition when forming the bond (default smaller, falling back to selected N atom if the two models contain the same numbers of atoms)
The selection must include exactly one atom from each model; these two atoms will be deleted and replaced with the new bond. Each of the selected atoms must be bonded to only one other atom, and it is these two other atoms that will form the new bond. Usually these atoms to be replaced are hydrogens, so it may be useful to add hydrogens beforehand, to whole models with Add Hydrogens or in a more local fashion with the Modify Structure section of Build Structure.
- Bond length – length for the new bond in Å, either entered directly or estimated from the covalent radii of the elements of the atoms to be bonded
- Set dihedral – allow specifying a Dihedral angle. This angle is defined by the newly bonded atoms (atom2, atom3) and their flanking bonds (atom1-atom2, atom3-atom4). If Set dihedral is used, the specifiers of atom2 and atom3 are shown in the dialog and pulldown menus on either side allow specifying which atoms to use as atom1 and atom4. If Set dihedral is not used, the two models will simply be aligned along the vectors of the two bonds to be replaced, with no attempt to rotate them to form a chemically reasonable dihedral angle.
- Move atoms in [ smaller / larger / #N ] model – which of the two models to reposition when forming the bond (default smaller)
Clicking Apply performs the join. If the new bond is between min-backbone atoms (both peptide or both nucleic), the residues from the moving model will be assigned the same chain ID as the rest of the chain in the nonmoving model, although their numbers may be shifted by a constant amount to avoid duplicate residue numbers within a chain. If the moving model includes additional chains, the other chain IDs will also be changed as needed to avoid duplicate chain IDs within a model (as described for combine).
The Invert section of Build Structure allows exchanging the positions of two substituents of an atom, potentially inverting a chiral center. Substituents of atoms that are not chiral centers can also be swapped.
Any unintended results can be reversed by clicking Swap again without changing the selection.
The Adjust Angles section of Build Structure is a table of active (currently adjustable) angles with entry fields and dials for interactive adjustment. Clicking Activate adds the angle defined by the three currently selected atoms to the list. Although the atoms need not be connected through consecutive bonds, there are other rules for which atoms can be used to define an active angle. Active angles are saved in sessions. See also: Angles/Torsions, angle, torsion
If the three atoms defining an active angle are 1-2-3, atom 1 is Fixed, atom 2 is Middle, and atom 3 is Moving, meaning that 3 and the side of the molecule bonded to it will move when the angle is adjusted. The angle can be adjusted by entering a new angle value or by manipulating the associated dial. Only values 0.0-180.0° are allowed.
The Middle column contains a pulldown menu labeled with the identifier of atom 2. This menu contains choices to:
The angle can be deactivated by clicking the “x” button to its left. To preserve the original position, make sure to reset the angle before deactivating it or swapping sides.
Approximate covalent bond radii are used to guess the connectivity of untemplated residues (when not specified in the input structure file) and to generate crude bond lengths for building atomic structures.
| Selected covalent bond radii (Å) | |
|---|---|
| H | 0.23 |
| B | 0.83 |
| C | 0.68 |
| N | 0.68 |
| O | 0.68 |
| F | 0.64 |
| Si | 1.20 |
| P | 1.05 |
| S | 1.02 |
| Cl | 0.99 |
| Se | 1.22 |
| Br | 1.21 |
| I | 1.40 |
A complete list, obtained many years ago from documentation from the Cambridge Crystallographic Data Centre, can be found in Table III of:
Determination of molecular topology and atomic hybridization states from heavy atom coordinates. Meng EC, Lewis RA. J Comput Chem 12:891 (1991).