November 6, 2006
Adds the Chimera command "sym" to show symmetry related copies of a molecule. As you move the original molecule relative to a density map the positions of all the symmetry copies are automatically updated.
The symmetry matrices are read from the REMARK 350 BIOMT records in the PDB file header. You can put these matrices in by hand with a text editor. Below is an example. The matrices represent the symmetry of the density map. It would make more sense to put them in the map file header but that is not as easy since it is a binary format file.
Once you open your PDB file with the BIOMT matrices in the header and display your density map type
sym
to the Chimera command-line (menu Favorites / Command Line). This will create copies of the molecule for each symmetry matrix (that is not the identity). If you freeze the density map by turning off the checkbutton in the Active column in Model Panel (menu Favorites / Model Panel) and then move the original molecule with the mouse, all symmetry copies will be moved using the BIOMT positioning matrices in the coordinate frame of the density map.
To remove the symmetry copies issue the command:
~sym
This is a Chimera convention that tilde in front of a command undoes the command.
Here are some more details. If you have multiple molecules opened in Chimera you can specify which one to use in the sym command using the model's id number listed in the Model Panel dialog. For example
sym #0
The density map shown in the volume viewer dialog will be used as the reference coordinate system for applying the symmetries. If you want to specify which map to use as the reference coordinate system give a second model id number:
sym #0 #1
If you are showing symmetry for multiple models you can remove the symmetry copies for just one molecule by giving its model id number with the ~sym command:
~sym #2
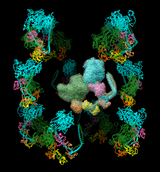
The image shows 12 copies of myosin arranged helically. The helical symmetry was specified by adding the following positioning matrices to PDB entry 1i84. Here is a movie showing one copy being moved. No density map is shown in this example.
REMARK 350 BIOMOLECULE: 1 REMARK 350 APPLY THE FOLLOWING TO CHAINS: S, T, U, V, W, Z REMARK 350 BIOMT1 1 1 0 0 0 REMARK 350 BIOMT2 1 0 1 0 0 REMARK 350 BIOMT3 1 0 0 1 0 REMARK 350 BIOMT1 2 0 -1 0 0 REMARK 350 BIOMT2 2 1 0 0 0 REMARK 350 BIOMT3 2 0 0 1 0 REMARK 350 BIOMT1 3 -1 0 0 0 REMARK 350 BIOMT2 3 0 -1 0 0 REMARK 350 BIOMT3 3 0 0 1 0 REMARK 350 BIOMT1 4 0 1 0 0 REMARK 350 BIOMT2 4 -1 0 0 0 REMARK 350 BIOMT3 4 0 0 1 0 REMARK 350 BIOMT1 5 0.866025 -0.5 0 0 REMARK 350 BIOMT2 5 0.5 0.866025 0 0 REMARK 350 BIOMT3 5 0 0 1 145 REMARK 350 BIOMT1 6 -0.5 -0.866025 0 0 REMARK 350 BIOMT2 6 0.866025 -0.5 0 0 REMARK 350 BIOMT3 6 0 0 1 145 REMARK 350 BIOMT1 7 -0.866025 0.5 0 0 REMARK 350 BIOMT2 7 -0.5 -0.866025 0 0 REMARK 350 BIOMT3 7 0 0 1 145 REMARK 350 BIOMT1 8 0.5 0.866025 0 0 REMARK 350 BIOMT2 8 -0.866025 0.5 0 0 REMARK 350 BIOMT3 8 0 0 1 145 REMARK 350 BIOMT1 9 0.866025 0.5 0 0 REMARK 350 BIOMT2 9 -0.5 0.866025 0 0 REMARK 350 BIOMT3 9 0 0 1 -145 REMARK 350 BIOMT1 10 -0.5 0.866025 0 0 REMARK 350 BIOMT2 10 -0.866025 -0.5 0 0 REMARK 350 BIOMT3 10 0 0 1 -145 REMARK 350 BIOMT1 11 -0.866025 -0.5 0 0 REMARK 350 BIOMT2 11 0.5 -0.866025 0 0 REMARK 350 BIOMT3 11 0 0 1 -145 REMARK 350 BIOMT1 12 0.5 -0.866025 0 0 REMARK 350 BIOMT2 12 0.866025 0.5 0 0 REMARK 350 BIOMT3 12 0 0 1 -145
Here is an annoying little problem. If you use the Unshow button on the volume dialog it removes the density map surface model and forgets its orientation. The symmetry copies of the molecule will no longer have their positions updated. Even if you redisplay the map by pressing the Show button, the symmetry repositioning will no longer work. You'll have to use ~sym followed by sym to get it working again. If you want to hide the map you can use the checkbutton in the Model Panel dialog to hide it without actually deleting the surface, so the symmetry copies will continue to have their positions updated.
Another problem to look for. Chimera can only handle about 100,000 atoms before it gets slow. Also it uses a few Kbytes of memory per atom. If you specify alot of symmetry matrices you can quickly create a large number of atoms. You may need to limit the symmetry matrices in the PDB header to keep the total number of atoms down.
November 6, 2006.
Fixed error that occured when sym command executed with no arguments and
PDB model has no BIOMT matrices.
September 21, 2005.
Developed sym command for Ed Egelman for docking actin into EM density maps.